




合作客戶/
拜耳公司 |
同濟大學 |
聯合大學 |
美國保潔 |
美國強生 |
瑞士羅氏 |






相關新聞Info
-
> 帶油涂裝涂料的優缺點、表面性能及研究
> 生物表面活性劑產生菌菌體密度、細胞疏水性與發酵液pH及表面張力的關系(二)
> 不同表面張力和接觸角下膨脹土裂隙的發展演化過程(三)
> 耐高溫采油菌株ZY-1:碳源對發酵液表面張力的影響
> 磷脂膜破裂臨界時間和臨界表面張力值分析方法
> 基于界面張力弛豫法探討疏水改性聚合物與石油酸和瀝青質間的相互作用(一)
> 基于表面張力方法判斷物質(或材料)的親水性(一)
> 基于深度神經網絡模型分析明膠溶液荷電量與表面張力之間的關系(二)
> 應用單分子層技術分析磷脂酶與不同磷脂底物特異水解性能:結果和討論、結論!
> 礦用塵克(C&C)系列除塵劑對大采高工作面截割煤塵的降塵效率影響(一)
推薦新聞Info
-
> Langmuir槽法研究不同電性Gemini表面活性劑對界面吸附膜性質的影響(三)
> Langmuir槽法研究不同電性Gemini表面活性劑對界面吸附膜性質的影響(二)
> Langmuir槽法研究不同電性Gemini表面活性劑對界面吸附膜性質的影響(一)
> 生物表面活性劑產生菌的篩選及對PAHs污染環境的修復效果研究(四)
> 生物表面活性劑產生菌的篩選及對PAHs污染環境的修復效果研究(三)
> 生物表面活性劑產生菌的篩選及對PAHs污染環境的修復效果研究(二)
> 生物表面活性劑產生菌的篩選及對PAHs污染環境的修復效果研究(一)
> 表面活性劑生物降解度測定方法種類及表面張力法的優勢——結果與分析、結論
> 表面活性劑生物降解度測定方法種類及表面張力法的優勢——摘要、實驗部分
> 炔屬二醇表面活性劑對環氧灌漿材料漿液性能、灌體的滲透性影響(二)
溫度對水—十二烷基硫酸鈉體系與純水體系界面張力、厚度的影響——模擬方法
來源:河南化工 瀏覽 373 次 發布時間:2025-04-14
摘要:采用分子動力學模擬技術,對水及其表面活性劑體系的汽—液界面行為進行了研究。模擬結果表明,隨著溫度的升高,純水體系液相主體密度降低,氣—液界面厚度增大,界面張力逐漸減小;水—十二烷基硫酸鈉體系與純水體系相比,汽—液界面厚度明顯增大,汽—液界面張力明顯減小,其隨溫度的變化規律和純水體系一致。
眾所周知,表面活性劑具有降低水的表面張力能力,其在氣—液界面上的吸附行為是發揮效用的關鍵。氣—液界面熱力學行為一直是相變傳熱傳質研究的重點。由于氣—液界面厚度非常薄,這就使得其理論分析和實驗研究變得十分困難。近些年來,隨著計算機技術的迅猛發展,越來越多的學者采用分子動力學(MD)模擬方法,來研究氣—液相變界面特性。Kuhn等采用分子動力學方法,考查了氣—液界面上的脂肪醇聚氧乙烯醚非離子表面活性劑(C12E5)單分子層的結構參數以及分子的動態行為。Wu等采用分子動力學模擬技術,分析了不同種類的胺基Gemini型表面活性劑在正庚烷—水體系的界面張力、密度分布,以及分子的微觀結構,其模擬結果與實驗吻合良好。苑世領等用分子動力學模擬的方法,研究了陰離子表面活性劑十二烷基硫酸鈉(SDS)在汽—液界面上的結構和動力學性質。肖紅艷等研究了不同油相和鹽度條件下表面活性劑—烷烴—水體系的界面結構,給出了徑向分布函數、二面角幾率變化等動力學結構信息。本文擬采用分子動力學模擬方法,利用LAMMPS軟件模擬水及其表面活性劑體系的氣—液界面行為。
1模擬方法
1.1模擬體系
采用直角坐標系,水體系的模擬盒子(初始狀態)如圖1所示,其大小為Lx×Ly×Lz=12 nm×4 nm×4 nm。液體水分子以面心立方(FCC)晶格方式排列于模擬盒子的中央,汽相分別處于液相的左右兩側,整個模擬體系中有兩個氣—液界面。

圖1水體系的模擬盒子(初始狀態)
采用直角坐標系,水—十二烷基硫酸鈉表面活性劑體系的模擬盒子(初始狀態)如圖2所示,其大小為Lx×Ly×Lz=12 nm×4 nm×4 nm。液體水分子以隨機分布的方式位于模擬盒子的中央,兩側各有一相對的表面活性劑單分子層,汽相分別處于液相的左右兩側,整個模擬體系中有兩個氣—液界面。

圖2水—十二烷基硫酸鈉體系的模擬盒子(初始狀態)
1.2勢能模型
水分子模型很多,如SPC、SPCE、TPI3P和TPI4P等,其結構示意圖和模型參數分別見圖3和表1。水分子的勢能函數如式(1)所示。
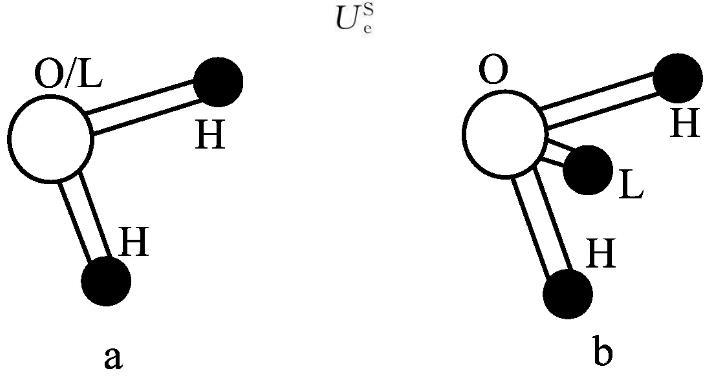
圖3不同水分子模型的結構示意圖
圖3a中為SPC、SPCE和TIP3P模型,b為TIP4P模型(L:負電荷作用點;H:正電荷作用點)
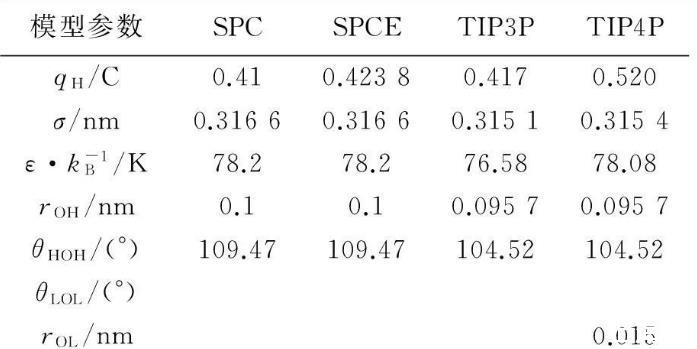
表1水分子模型參數
表中:q,電量,C;σ,尺度參數,nm;ε,能量參數,J;kB,玻爾茲曼常數,J/K;r,分子間距,nm;θ鍵角,(°)。
在水—表面活性劑體系的MD模擬中,十二烷基硫酸鈉采用全原子模型,力場參數基于AMBER力場,其函數形式如方程(2)所示。
式中:kr、kθ、Vn分別為鍵力常數、彎曲力常數、二面角扭曲常數;l0、θ0分別為標準鍵長和標準鍵角;n為整數(繞鍵旋轉360°時出現的能量最小值的數目);φ為二面角;rij為原子i和j之間的距離;靜電相互作用項中的q表示原子上的電荷數,e。不同原子間的范德華相互作用項中的εij和σij,采用Lorentz-Berthelot混合規則。
1.3模擬細節
水體系模擬在x、y、z方向均采用周期性邊界條件,原子間力的截斷半徑為12 nm,模擬時間步長為1 fs,總模擬時間為0.6 ns,前0.4 ns使得系統達到平衡,后0.2 ns統計計算并輸出系統的密度分布、界面張力以及界面厚度。采取正則系綜(NVT),并采用Woodcock控溫法維持體系溫度衡定;依照設定的溫度,隨機分布分子的初始平動速度;為了保證水分子不偏離盒子中心,每隔1 000步矯正體系的質心,使之在x、y、z方向始終處于盒子的中心處;水—十二烷基硫酸鈉體系模擬原子間力的截斷半徑為10 nm,庫倫力的截斷半徑為12 nm;模擬時間步長為1 fs,總模擬時間為1.4 ns,前1.0 ns使得系統達到平衡,后0.4 ns統計計算并輸出數據,其他的模擬設置同水體系一樣。本文模擬數據均采用LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)軟件計算得到。